|
|

|

|
| e-Pub |
Section: New Results
Molecular Modeling
Rapid determination of RMSDs corresponding to macromolecular rigid body motions
Participants : Petr Popov, Sergei Grudinin.
Finding the root mean sum of squared deviations (RMSDs) between two coordinate vectors that correspond to the rigid body motion of a macromolecule is an important problem in structural bioinformatics, computational chemistry and molecular modeling. Standard algorithms compute the RMSD with time proportional to the number of atoms in the molecule. We developed RigidRMSD, a new algorithm that determines a set of RMSDs corresponding to a set of rigid body motions of a macromolecule in constant time with respect to the number of atoms in a molecule. Our algorithm is particularly useful for rigid body modeling applications such as rigid body docking, and also for high-throughput analysis of rigid body modeling and simulation results. A C++ implementation of our algorithm will be available at http://nano-d.inrialpes.fr/software/RigidRMSD .
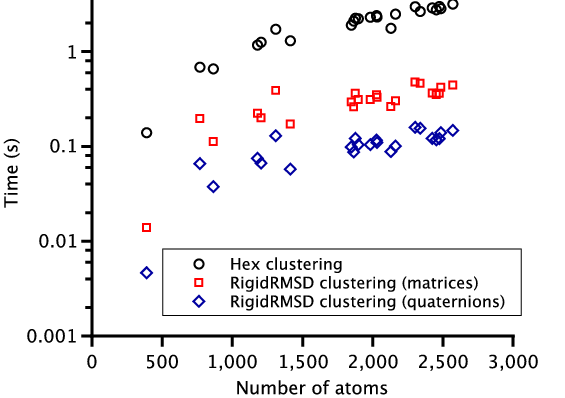
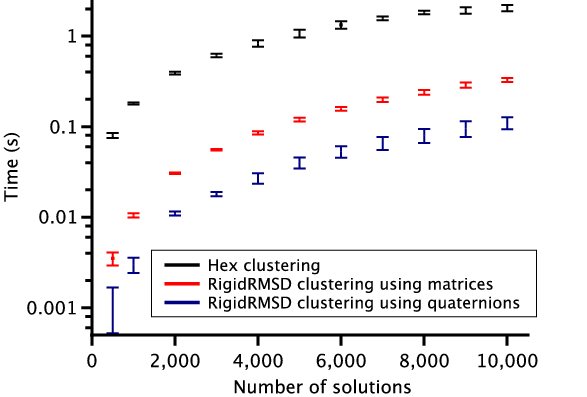
To demonstrate the efficiency of the RigidRMSD library, we compared the clustering application implemented with our algorithm to the one from the Hex software. We chose Hex for the comparison because it is a very fast rigid body docking tool and also because it explicitly provides the clustering time. For the comparison, we collected a small benchmark of 23 protein dimers of various size. After, we launched Hex version 6.3 on this benchmark and collected docking solutions before clustering, sizes of clusters, and clustering time. We then also clustered these solutions using the RigidRMSD library. Figure 6 shows the clustering time of the HEX clustering algorithm with respect to our clustering using two rotation representations as a function of the number of atoms in the smaller protein (left) and the number of docking solutions before the clustering (right). We can clearly see that our implementation of the clustering algorithm is more than an order of magnitude faster compared to the Hex implementation. Also, the quaternion representation of rotation is on average three times more efficient compared to the matrix representation.
|
Fast fitting atomic structures into a low-resolution density map using 3D orthogonal Hermite functions
Participants : Georgy Derevyanko, Sergei Grudinin.
We developed a new algorithm for fitting protein structures into a low resolution electron density (e.g. cryo-electron microscopy) map. The algorithm uses 3D orthogonal Hermite functions for fast operations on the electron density.
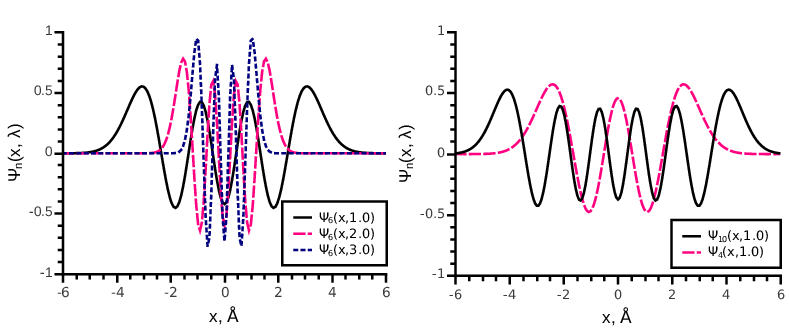
Orthogonal Hermite function of order is defined as:
where is the Hermite polynomial and is the scaling parameter. In Fig. 7 we show several orthogonal Hermite functions of different orders with different parameters . These functions form an orthonormal basis set in . A 1D function decomposed into the set of 1D Hermite functions up to an order reads
Here, are the decomposition coefficients, which can be determined from the orthogonality of the basis functions . Decomposition in Eq. 2 is called the band-limited decomposition with basis functions. To decompose the electron density map and the protein structures, we employ the 3D Hermite functions:
which form an orthonormal basis set in . A function represented as a band-limited expansion in this basis reads
|
Our algorithm accelerates rotation of the Fourier image of the electron density by using 3D orthogonal Hermite functions. As a part of the new method, we presented an algorithm for the rotation of the density in the Hermite basis and an algorithm for the conversion of the expansion coefficients into the Fourier basis. We implemented the program of fitting a protein structure to a low-resolution electron density map, which uses the cross-correlation or the Laplacian-filtered cross-correlation as the fitting criterion. We demonstrated that in the Hermite basis, the Laplacian filter has a particularly simple form. To assess the quality of density encoding in the Hermite basis, we uses two measures, the R-factor and the cross-correlation factor. Finally, we validated our algorithm using two examples and compare its efficiency with two widely used fitting methods, ADP EM and colores from the Situs package.
Fast Rotational-Translation Matching of Rigid Bodies by Fast Fourier Transform Acceleration of Six Degrees of Freedom
Participants : Alexandre Hoffmann, Sergei Grudinin.
We introduced a new method for rigid molecular fitting. This problem is usually solved by maximizing the Cross Corelation Function (CCF), which is computed using the Fast Fourier Transform (FFT) algorithm. Our method handles six degrees of freedom at once and requires only one computation of the Cross Corelation Function, with the six-dimensional Fast Fourier Transform. Our method only requires a low pre-processing time (), which is comparable to the cost of the subsequent 6D FFT (). It also uses a dual Hermite-Fourier representation, which allows to represent a small molecule with a fewer number of coefficients in the Hermite basis.
Prediction of complexes with point group symmetry using spherical polar Fourier docking correlations
Participants : David W. Ritchie, Sergei Grudinin.
Many proteins form symmetric homo-oligomers that perform a certain physiological function. We present the first point group symmetry docking algorithm that generates perfectly symmetrical protein complexes for arbitrary point group symmetry types (, , , , and ). We validate the algorithm on proteins from the 3D-Complex database, where it achieves on average the success rate of 55%. The running time of the algorithm is less than a minute on a modern workstation.
Many of the protein complexes in the protein Data bank (PDB) are symmetric homo-oligomers. According to the 3D-Complex database, homo-dimers comprise the majority of known homo-oligomers. However, many complexes have higher order rotational symmetry (i.e. >2), and a good number have multiple rotational symmetry axes, namely those with dihedral (), tetrahedral (), octahedral (), and icosahedral () point group symmetries. Although symmetrical complexes are often solved directly by X-ray crystallography, it would still be very useful to be able to predict whether or not a given monomer might self-assemble into a symmetrical structure. We present a new point group symmetry docking algorithm. In the last few years, several protein-protein docking programs have been adapted to predict symmetrical pair-wise docking orientations for and symmetries. However, to our knowledge, there does not yet exist an algorithm which can automatically generate perfectly symmetrical protein complexes for arbitrary point group symmetry types.
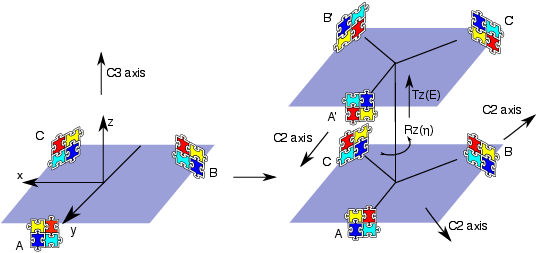
We introduce the notion of a "docking equation" in which the notation represents an interaction between proteins and in 3D space. It is also useful introduce the operators and , which represent the actions of translating an object by an amount and rotating it according to the three Euler rotation angles . Then, guided by Figure 8 , and assuming that we start with two identical monomers at the origin, we can write down a docking equation for the two monomers as
Then, we perform a series of fast Fourier transform (FFT) correlation searches using the Hex spherical polar Fourier docking algorithm to determine the four parameters . For higher symmetries, , , , and , we introduce two more parameters and perform a series of FFT in a similar way. The calculation for each structure takes less than a minute on a modern workstation.
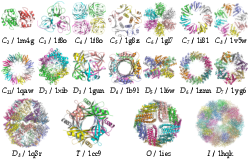
We validated our method on protein structures from the 3D-Complex database, which contains 17,183 protein complexes with assigned biological unit and symmetry type. It mostly contains cyclic and dihedral proteins, as well as 86 tetrahedral, 47 octahedral, and 6 icosahedral complexes (excluding all viral structures). Starting from the structures of monomers, we generated symmetric biological units based on the symmetry type for each complex provided by 3D-Complex. Figure 9 summarizes the performance of our method on these proteins, where we say that the model is correct if all pair-wise RMSDs are smaller than 10 Ångstroms. On average, we found about 55% of correct predictions ranked first.
|
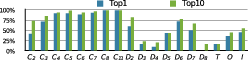
Figure 10 shows correctly predicted examples from each of the symmetry types. Each complex is perfectly symmetrical, although due to the soft docking function in Hex it is possible that some interfaces might contain minor steric clashes.